ARUP Laboratories To Host Informational Webinar on the FDA’s Rule To Regulate Lab-Developed Tests
ARUP Laboratories will host an informational webinar on May 23, 2024, at 10 a.m., for clients, members of the media, and others to discuss the FDA’s rule to regulate laboratory-developed tests (LDTs) as medical devices. Jonathan Genzen, MD, PhD, ARUP’s chief medical officer and senior director of governmental affairs, and Jonathan Carr, JD, chief compliance officer, will summarize the details of the final rule and discuss the anticipated challenges for labs, patients, and providers.

The FDA released the final rule on April 29, 2024, without incorporating many concerns raised by ARUP Laboratories and other members of the clinical laboratory community. ARUP’s stance on the rule has not changed, CEO Andy Theurer said. ARUP believes the FDA does not have statutory authority over LDTs and maintains that the rule will limit access to essential testing services, stifle innovation, and increase healthcare costs.
“ARUP is evaluating and reviewing the final rule, including any changes, internally and with our industry partners,” Theurer said. “Our focus remains on supporting our patients and our clients to ensure they do not lose access to the essential services we provide.”
“ARUP is committed to maintaining our extensive test menu and advocating at the national level on behalf of the clinical laboratory community and the patients we serve,” Genzen added.
The final rule differs from the FDA’s proposed rule published in October in that it permits enforcement discretion in some instances, such as for LDTs currently on the market and for those approved by the New York State Clinical Laboratory Evaluation Program (CLEP). Labs will have to comply with new record-keeping and labeling requirements for many of those tests.
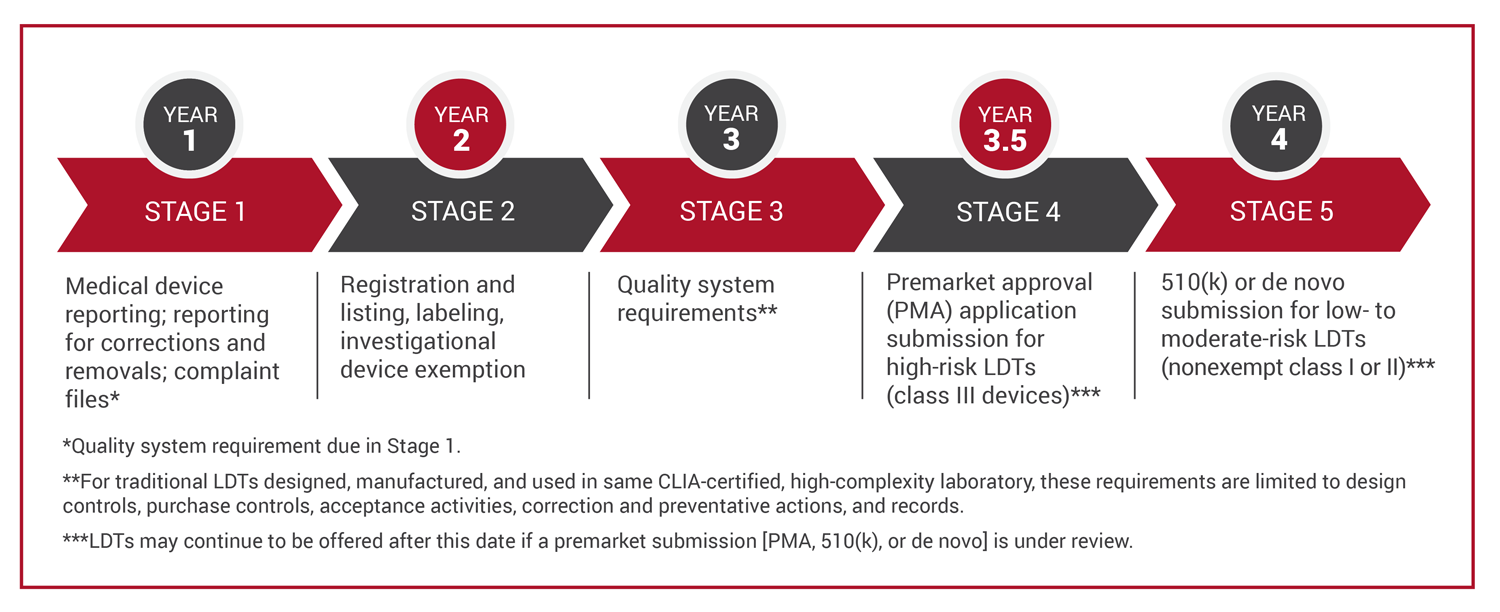
The rule will be phased in over the next four years. In the first year, by May 6, 2025, laboratories that offer LDTs will be required to file medical device reports with the FDA. Year two adds requirements for registering and listing LDTs with the FDA. By year three, clinical laboratories will need to begin applying FDA quality system requirements (QSRs), and by year three and a half, clinical laboratories will be required to submit premarket approvals for high-risk LDTs. Current FDA user fees can be as high as $483,000 for each premarket authorization submission. The final stage, at year 4, adds premarket notification requirements for low- and moderate-risk LDTs.
In a November 28, 2023, public comment on the FDA’s proposed rule, ARUP urged the FDA to withdraw the proposal, citing negative impacts to patient care and innovation. (ARUP’s public comment is linked here.) ARUP believes the FDA used exaggerated estimates of the number of LDTs ordered and flawed data about their performance. LDTs address a vital need and have an excellent performance record in ARUP’s laboratories and in external proficiency testing.
ARUP also challenged the agency’s regulatory authority over laboratory-developed testing services. Congress gave the FDA authority to regulate medical devices, but if Congress had intended the FDA to regulate laboratory-developed testing services as medical devices, lawmakers would have included this concept in the text of the Medical Device Amendments of 1976 (MDA), and did not.
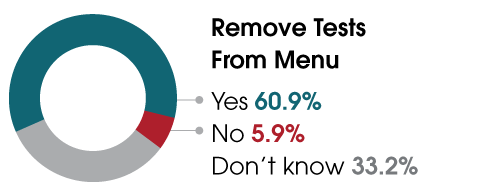
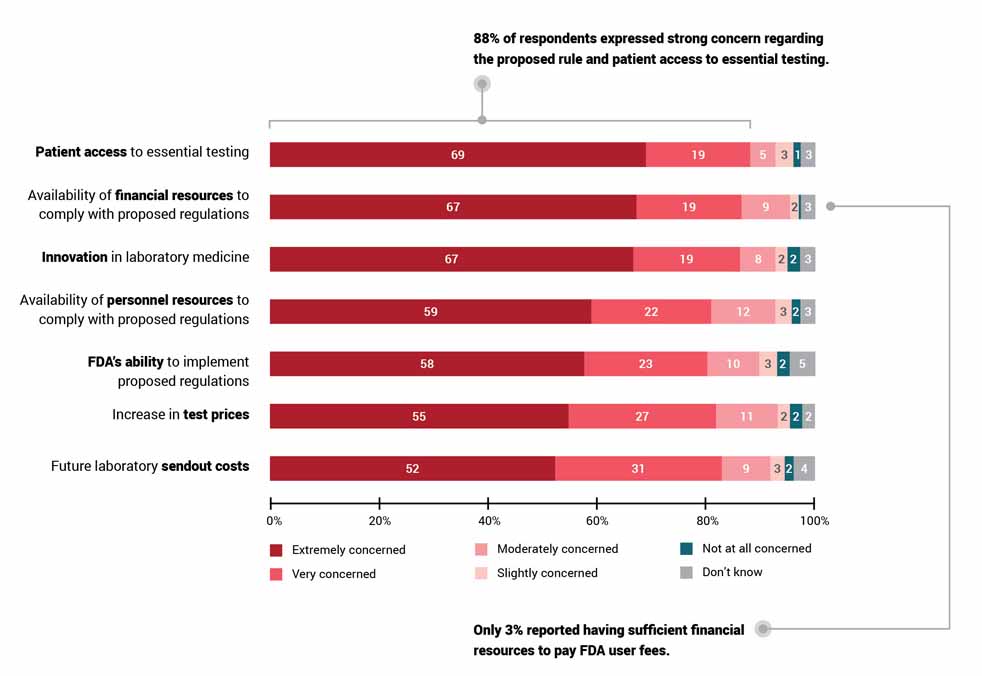
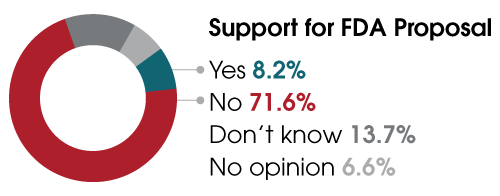
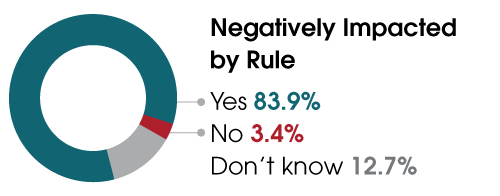
This past spring, ARUP conducted the largest survey to date assessing the impact of the proposed rule on clinical laboratories. Nearly 85% of respondents indicated the rule would hurt their laboratories and, ultimately, patient care. Of the 503 respondents, only 41 (8.2%) supported the FDA’s proposal, and of those whose labs perform LDTs, more than 60% said they would have to remove tests from their menus. A third indicated they did not know if they would need to remove tests. ARUP’s upcoming webinar aims to educate industry professionals so that labs can determine their next steps.
As an alternative to FDA regulation of LDTs, ARUP has advocated for a collaborative process to update existing regulations that now govern clinical laboratories under the Centers for Medicare and Medicaid Services Clinical Laboratory Improvement Amendments. Genzen said a collaborative legislative approach would be a better option.
“We are disappointed that the FDA enacted the rule, denying many stakeholders the opportunity to collaborate on an oversight framework aligned with clinical laboratory expertise and resources in order to better protect patients and future innovation,” Genzen said.
ARUP Laboratories is asking participants to register in advance for the webinar and to submit questions to help shape the conversation to address attendees’ main concerns.
- Register for the webinar here.
- Read ARUP’s statement in response to the final rule here.
- Explore ARUP’s survey of the lab community’s sentiments regarding the rule here.
Webinar To Discuss the FDA’s Rule To Regulate Laboratory-Developed Tests (LDTs) as Medical Devices
ARUP Laboratories will host a webinar on May 23, 2024, at 10 a.m., to summarize the details of the final rule and discuss the anticipated challenges for labs, patients, and providers.
Register for the webinar here.
ARUP Laboratories Survey
Lab Community “Extremely Concerned” About Patient Access to Testing If FDA Regulates Lab-Developed Tests

Nearly 85% of respondents to an ARUP Laboratories survey on the impact of the FDA’s proposed rule to regulate laboratory-developed tests (LDTs) believe the proposal will hurt their laboratories and, ultimately, patient care.
Survey Results
Of 503 respondents, only 8% support the FDA's proposed rule.
Of respondents whose laboratories perform LDTs, nearly 84% believe the rule will negatively impact their laboratories.
The majority of laboratories believe they will have to remove tests from their menus.